GAM-clustering reanalysis of ImmGen Open Source bulk RNA-Seq data
Source:vignettes/GAMclust_tutorial_BULK.Rmd
GAMclust_tutorial_BULK.RmdInstall GAMclust package:
devtools::install_github("alserglab/GAMclust")
library(GAMclust)
library(gatom)
library(mwcsr)
library(fgsea)
library(data.table)
library(futile.logger)
set.seed(42)
startTime <- Sys.time()Preparing working environment
First, please load and initialize all objects required for GAM-clustering analysis:
- load metabolic network and its metabolites annotation. We provide two networks: KEGG and combined network that includes KEGG, Rhea and transport reactions:
(1.1.) load KEGG metabolic
network network.kegg.rds
and its metabolites annotation met.kegg.db.rds
# KEGG network:
network <- readRDS(url("http://artyomovlab.wustl.edu/publications/supp_materials/GATOM/network.kegg.rds"))
metabolites.annotation <- readRDS(url("http://artyomovlab.wustl.edu/publications/supp_materials/GATOM/met.kegg.db.rds"))(1.2.) or load combined metabolic network network.combined.rds,
its metabolites annotation met.combined.db.rds
and species-specific list of genes that either come from proteome or are
not linked to a specific enzyme gene2reaction.combined.mmu.eg.tsv
for mouse and gene2reaction.combined.hsa.eg.tsv
for human data;
# combined network (KEGG+Rhea+transport reactions):
network <- readRDS(url("http://artyomovlab.wustl.edu/publications/supp_materials/GATOM/network.combined.rds"))
metabolites.annotation <- readRDS(url("http://artyomovlab.wustl.edu/publications/supp_materials/GATOM/met.combined.db.rds"))
gene2reaction.extra <- data.table::fread("http://artyomovlab.wustl.edu/publications/supp_materials/GATOM/gene2reaction.combined.mmu.eg.tsv", colClasses="character")- load species-specific network annotation:
org.Hs.eg.gatom.annofor human data ororg.Mm.eg.gatom.annofor mouse data;
network.annotation <- readRDS(url("http://artyomovlab.wustl.edu/publications/supp_materials/GATOM/org.Mm.eg.gatom.anno.rds"))- load provided list of metabolites that should not be considered during the analysis as connections between reactions (e.g., CO2, HCO3-, etc);
met.to.filter <- data.table::fread(system.file("mets2mask.lst", package="GAMclust"))$ID- initialize SMGWCS solver:
(4.1.) we recommend to use here either heuristic relax-and-cut solver
rnc_solver from mwcsr
package,
solver <- mwcsr::rnc_solver()(4.2.) either proprietary CPLEX solver (free for academy);
cplex.dir <- "/opt/ibm/ILOG/CPLEX_Studio1271"
solver <- mwcsr::virgo_solver(cplex_dir = cplex.dir)- set working directory where the results will be saved to.
work.dir <- "results-bulk"
dir.create(work.dir, showWarnings = F, recursive = T)- TEMPORARY: collecting logs while developing the tool.
stats.dir <- file.path(work.dir, "stats")
dir.create(stats.dir, showWarnings = F, recursive = T)
setup_logger <- function(log.file.path, logger.name = "stats.logger") {
file.appender <- appender.file(log.file.path)
console.appender <- appender.console()
combined.appender <- function(line) {
file.appender(line)
console.appender(line)
}
flog.appender(combined.appender, name = logger.name)
flog.threshold(TRACE, name = logger.name)
}
log.file <- file.path(stats.dir, "log.txt")
setup_logger(log.file.path = log.file, logger.name = "stats.logger")Preparing objects for the analysis
Preparing data
GAMclust works with bulk, single cell and spatial RNA-seq data.
This vignette shows how to process bulk RNA-seq data on the example of ImmGen Open Source data reanalysis.
Let’s load the data.
For bulk RNA-seq cell data, take 12,000-15,000 genes for the GAM-clustering analysis.
expression_set_object <- readRDS(url("http://artyomovlab.wustl.edu/publications/supp_materials/GAMclust/243_es.top12k.rds"))
E <- Biobase::exprs(expression_set_object)
nrow(E) # ! make sure this value is in range from 10,000 to 15,000# [1] 12000
E[1:3, 1:3]# MF.64pLYVEpIIn.Ao.1 MF.64pLYVEpIIn.Ao.2 MF.64pLYVEpIIn.Ao.3
# Actb 14.77450 14.97466 14.80920
# Cst3 12.73106 12.80513 12.62843
# Eef1a1 12.14864 12.38202 12.33756Genes in your dataset may be named as Symbol, Entrez, Ensembl or
RefSeq IDs. One of these names should be specified as value of
gene.id.type parameter in prepareData().
E.prep <- prepareData(E = E,
gene.id.type = "Symbol",
use.PCA = FALSE,
network.annotation = network.annotation)
E.prep[1:3, 1:3]# MF.64pLYVEpIIn.Ao.1 MF.64pLYVEpIIn.Ao.2 MF.64pLYVEpIIn.Ao.3
# 11461 0.7947992 1.0375496 0.8368845
# 13010 0.3038730 0.3471841 0.2438681
# 13627 0.4187557 0.6674637 0.6200901Preparing network
The prepareNetwork() function defines the structure of
the final metabolic modules.
network.prep <- prepareNetwork(E = E.prep,
network = network,
topology = "metabolites",
met.to.filter = met.to.filter,
network.annotation = network.annotation,
gene2reaction.extra = gene2reaction.extra) # for combined network# INFO [2026-07-06 16:01:06] Global metabolite network contains 6583 edges.# INFO [2026-07-06 16:01:06] Largest connected component of this global network contains 1430 nodes and 5121 edges.Preclustering
The preClustering() function defines initial patterns
using k-medoids clustering on gene expression matrix. It is strongly
recommended to do initial clustering with no less than 32 clusters
(initial.number.of.clusters = 32).
You can visualize the initial heatmap as shown below.
cur.centers <- preClustering(E.prep = E.prep,
network.prep = network.prep,
initial.number.of.clusters = 32,
network.annotation = network.annotation)# INFO [2026-07-06 16:01:06] 1160 metabolic genes from the analysed dataset mapped to this component.
cur.centers[1:3, 1:3]# MF.64pLYVEpIIn.Ao.1 MF.64pLYVEpIIn.Ao.2 MF.64pLYVEpIIn.Ao.3
# 1 -0.09284514 -0.12763246 0.02188235
# 2 0.03432400 0.02773624 0.13477218
# 3 0.78988889 0.80944803 0.87514519
pheatmap::pheatmap(
GAMclust:::normalize.rows(cur.centers),
cluster_rows=F, cluster_cols=F,
show_rownames=F, show_colnames=T)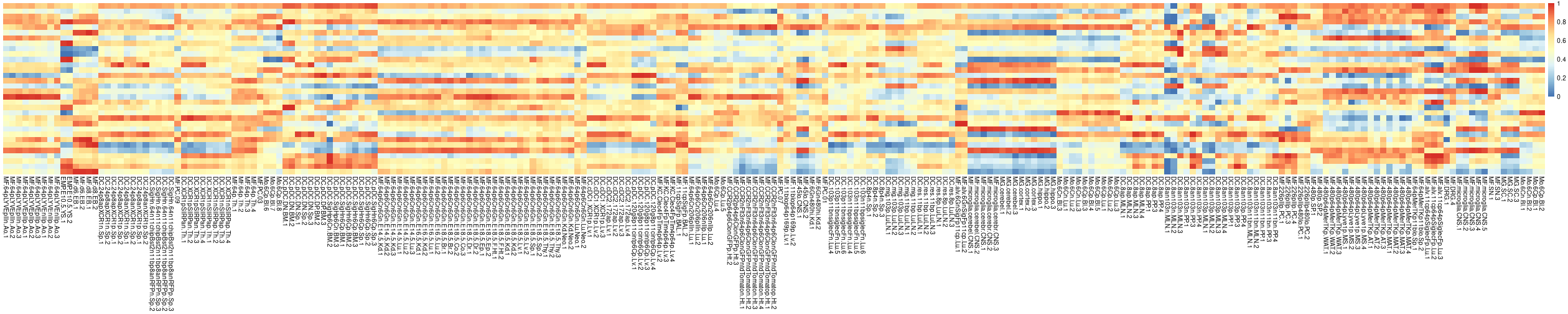
GAM-clustering analysis
Now you have everything prepared for the GAM-clustering analysis.
Initial patterns will be now refined in an iterative process. The
output of gamClustering() function presents a set of
specific subnetworks (also called metabolic modules) that reflect
metabolic variability within a given transcriptional dataset.
Note, that it may take a long time to derive metabolic modules by
gamClustering() function (tens of minutes).
There is a set of parameters which determine the size and number of your final modules. We recommend you to start with the default settings, however you can adjust them based on your own preferences:
If you consider final modules to be too small or too big and it complicates interpretation for you, you can either increase or reduce by 10 units the
max.module.sizeparameter.If among final modules you consider presence of any modules with too similar patterns, you can reduce by 0.1 units the
cor.thresholdparameter.If among final modules you consider presence of any uninformative modules, you can reduce by 10 times the
p.adj.val.thresholdparameter.
results <- gamClustering(E.prep = E.prep,
network.prep = network.prep,
cur.centers = cur.centers,
start.base = 0.5,
base.dec = 0.1,
max.module.size = 50,
cor.threshold = 0.7,
p.adj.val.threshold = 1e-5,
batch.solver = seq_batch_solver(solver),
work.dir = work.dir,
show.intermediate.clustering = F,
verbose = T,
collect.stats = T)Visualizing and exploring the GAM-clustering results
Each metabolic module is a connected piece of metabolic network whose genes expression is correlated across all dataset.
The following functions will help you to visualize and explore the obtained results.
Get graphs of modules:
getGraphs(modules = results$modules,
network.annotation = network.annotation,
metabolites.annotation = metabolites.annotation,
seed.for.layout = 42,
work.dir = work.dir)# Graphs for module 1 are built# Graphs for module 2 are built# Graphs for module 3 are built# Graphs for module 4 are built# Graphs for module 5 are built# Graphs for module 6 are built# Graphs for module 7 are built# Graphs for module 8 are built# Graphs for module 9 are builtExample of the graph of the first module:

Get gene tables:
The table contains gene list. Each gene has two descriptive values: i) gene’s correlation value with the modules pattern and ii) gene’s score. High score means that this gene’s expression is similar to the module’s pattern and not similar to other modules’ patterns.
m.gene.list <- getGeneTables(modules = results$modules,
nets = results$nets,
patterns = results$patterns.pos,
gene.exprs = E.prep,
network.annotation = network.annotation,
work.dir = work.dir)# Gene tables for module 1 are produced# Gene tables for module 2 are produced# Gene tables for module 3 are produced# Gene tables for module 4 are produced# Gene tables for module 5 are produced# Gene tables for module 6 are produced# Gene tables for module 7 are produced# Gene tables for module 8 are produced# Gene tables for module 9 are producedExample of the gene table of the first module:
head(GAMclust:::read.tsv(file.path(work.dir, "m.1.genes.tsv"))) |>
kableExtra::kable() |>
kableExtra::kable_styling()| symbol | Entrez | score | cor |
|---|---|---|---|
| Plod1 | 18822 | 2.211137 | 0.9426139 |
| Gbgt1 | 227671 | 1.545965 | 0.9089996 |
| Pla2g15 | 192654 | 1.501044 | 0.9061216 |
| Oplah | 75475 | 1.120957 | 0.8778247 |
| Ptgs1 | 19224 | 1.081628 | 0.8744483 |
| Slc2a8 | 56017 | 1.042379 | 0.8709858 |
Get plots of patterns and individual genes expression:
Heatmap for patterns of all modules:
patterns <- results$patterns.pos
pheatmap::pheatmap(
GAMclust:::normalize.rows(patterns),
cluster_rows=F, cluster_cols=F,
show_rownames=T, show_colnames=T)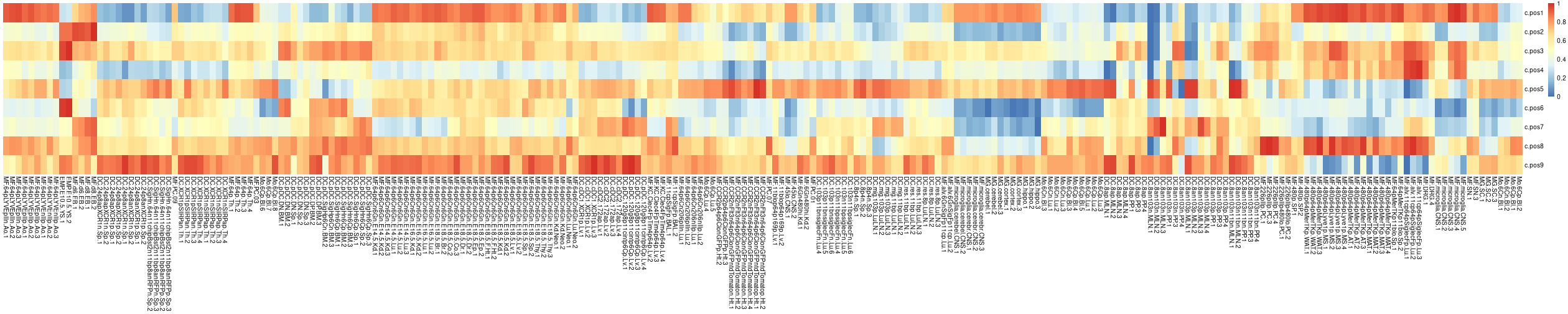
Get plots of individual genes expression
for (i in seq_along(m.gene.list)) {
heatmap <- E[m.gene.list[[i]], , drop=F]
pheatmap::pheatmap(
GAMclust:::normalize.rows(heatmap),
cluster_rows=F, cluster_cols=F,
file=sprintf("%s/m.%s.genes.png", work.dir, i),
width=10, height=5,
show_rownames=T, show_colnames=T)
}Example of the heatmap of the first module:
pheatmap::pheatmap(
GAMclust:::normalize.rows(E[m.gene.list[[1]], , drop=F]),
show_rownames=T, show_colnames=T)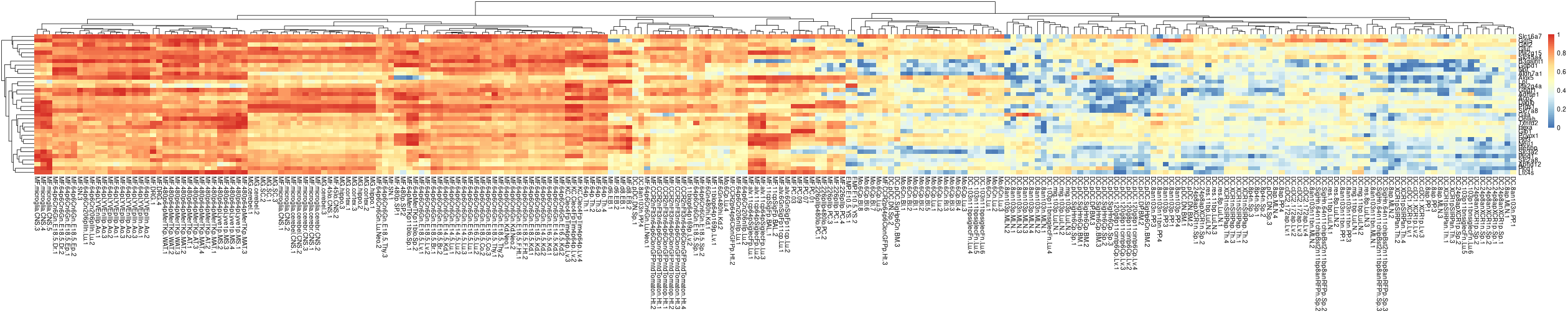
Get annotation tables and annotation heatmap for all modules:
Functional annotation of obtained modules is done based on KEGG and Reactome canonical metabolic pathways.
getAnnotationTables(network.annotation = network.annotation,
nets = results$nets,
work.dir = work.dir)# Pathway annotation for module 1 is produced# Pathway annotation for module 2 is produced# Pathway annotation for module 3 is produced# Pathway annotation for module 4 is produced# Pathway annotation for module 5 is produced# Pathway annotation for module 6 is produced# Pathway annotation for module 7 is produced# Pathway annotation for module 8 is produced# Pathway annotation for module 9 is producedExample of the annotation table of the first module:
head(GAMclust:::read.tsv(file.path(work.dir, "m.1.pathways.tsv"))) |>
kableExtra::kable() |>
kableExtra::kable_styling(font_size = 8) |>
kableExtra::column_spec(1, width = "1.6in") |>
kableExtra::column_spec(2:6, width = "0.5in") |>
kableExtra::column_spec(7, width = "1.2in")| pathway | pval | padj | foldEnrichment | overlap | size | overlapGenes |
|---|---|---|---|---|---|---|
| mmu00590: Arachidonic acid metabolism | 0.0000713 | 0.0170328 | 9.250000 | 5 | 10 | Alox5 Ltc4s Pla2g4a Ptgs1 Ggt5 |
| mmu00511: Other glycan degradation | 0.0025885 | 0.2262868 | 9.250000 | 3 | 6 | Glb1 Hexa Neu1 |
| mmu00603: Glycosphingolipid biosynthesis - globo and isoglobo series | 0.0043623 | 0.2606492 | 7.928571 | 3 | 7 | Hexa Gbgt1 B3galnt1 |
| mmu00565: Ether lipid metabolism | 0.0097110 | 0.4641838 | 6.166667 | 3 | 9 | Pla2g4a Pld3 Gdpd1 |
| mmu00220: Arginine biosynthesis | 0.0502266 | 1.0000000 | 5.285714 | 2 | 7 | Gpt2 Glul |
Annotation heatmap for all modules:
getAnnotationHeatmap(work.dir = work.dir)# Processing module 1# Module size: 34# Processing module 2# Module size: 19# Processing module 3# Module size: 10# Processing module 4# Module size: 12# Processing module 5# Module size: 7# Processing module 6# Module size: 5# Processing module 7# Module size: 5# Processing module 8# Module size: 5# Processing module 9# Module size: 6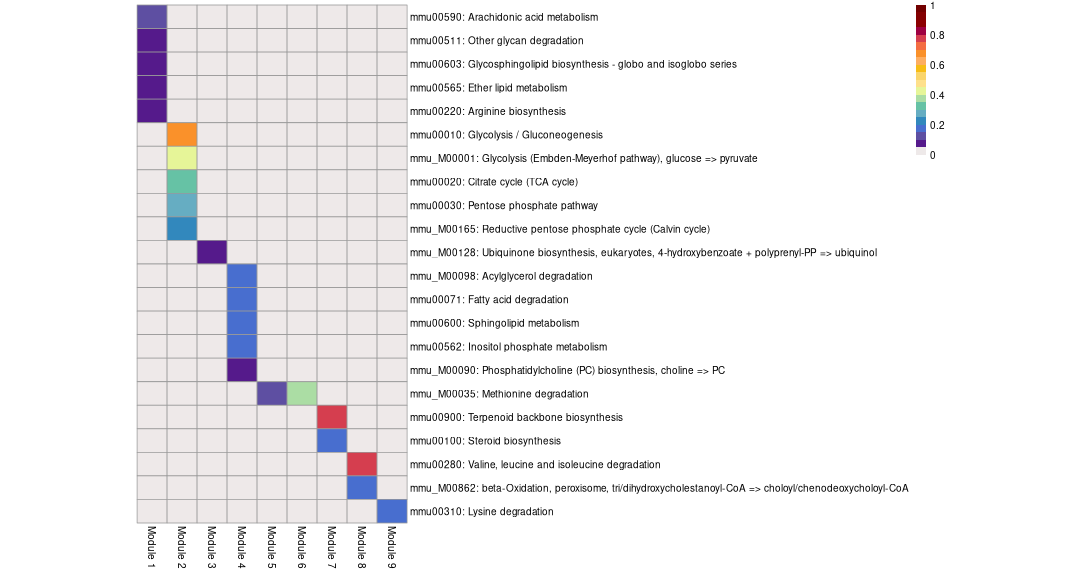
Compare modules obtained in different runs:
You may also compare two results of running GAM-clustering on the
same dataset (e.g. runs with different parameters) or compare two
results of running GAM-clustering on different datasets (then set
same.data=FALSE).
modulesSimilarity(dir1 = work.dir,
dir2 = "old_dir",
name1 = "new",
name2 = "old",
same.data = TRUE,
use.genes.with.pos.score = TRUE,
work.dir = work.dir,
file.name = "comparison.png")Session info
Elapsed time: 41.1 mins.
Peak R memory usage: 1.0 GB
# R version 4.5.3 (2026-03-11)
# Platform: x86_64-pc-linux-gnu
# Running under: Debian GNU/Linux 13 (trixie)
#
# Matrix products: default
# BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
# LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.29.so; LAPACK version 3.12.0
#
# locale:
# [1] LC_CTYPE=C.utf8 LC_NUMERIC=C LC_TIME=C
# [4] LC_COLLATE=C.utf8 LC_MONETARY=C.utf8 LC_MESSAGES=C.utf8
# [7] LC_PAPER=C.utf8 LC_NAME=C LC_ADDRESS=C
# [10] LC_TELEPHONE=C LC_MEASUREMENT=C.utf8 LC_IDENTIFICATION=C
#
# time zone: America/Chicago
# tzcode source: system (glibc)
#
# attached base packages:
# [1] stats graphics grDevices utils datasets methods base
#
# other attached packages:
# [1] futile.logger_1.4.9 fgsea_1.37.4 mwcsr_0.1.12
# [4] gatom_1.8.4 GAMclust_0.1.0 data.table_1.18.4
# [7] rmarkdown_2.31
#
# loaded via a namespace (and not attached):
# [1] tidyselect_1.2.1 viridisLite_0.4.3 dplyr_1.2.1
# [4] farver_2.1.2 blob_1.3.0 Biostrings_2.78.0
# [7] S7_0.2.2 fastmap_1.2.0 GGally_2.4.0
# [10] XML_3.99-0.23 digest_0.6.39 lifecycle_1.0.5
# [13] rsvg_2.7.0 KEGGREST_1.53.4 RSQLite_3.52.0
# [16] magrittr_2.0.5 compiler_4.5.3 rlang_1.2.0
# [19] tools_4.5.3 igraph_2.3.2 knitr_1.51
# [22] lambda.r_1.2.4 htmlwidgets_1.6.4 bit_4.6.0
# [25] xml2_1.5.2 plyr_1.8.9 RColorBrewer_1.1-3
# [28] BiocParallel_1.44.0 purrr_1.2.2 BiocGenerics_0.56.0
# [31] shinyCyJS_1.0.0 grid_4.5.3 stats4_4.5.3
# [34] ggplot2_4.0.3 scales_1.4.0 cli_3.6.6
# [37] crayon_1.5.3 generics_0.1.4 otel_0.2.0
# [40] rstudioapi_0.17.1 httr_1.4.8 DBI_1.3.0
# [43] cachem_1.1.0 stringr_1.6.0 parallel_4.5.3
# [46] AnnotationDbi_1.72.0 formatR_1.14 XVector_0.50.0
# [49] matrixStats_1.5.0 vctrs_0.7.3 Matrix_1.7-4
# [52] IRanges_2.44.0 S4Vectors_0.48.1 bit64_4.8.0
# [55] BioNet_1.70.0 Rgraphviz_2.54.0 systemfonts_1.3.1
# [58] tidyr_1.3.2 glue_1.8.1 ggstats_0.13.0
# [61] codetools_0.2-20 cowplot_1.2.0 stringi_1.8.7
# [64] gtable_0.3.6 tibble_3.3.1 pillar_1.11.1
# [67] htmltools_0.5.9 Seqinfo_1.0.0 graph_1.88.1
# [70] R6_2.6.1 textshaping_1.0.4 evaluate_1.0.5
# [73] kableExtra_1.4.0 Biobase_2.70.0 lattice_0.22-7
# [76] futile.options_1.0.1 png_0.1-9 pheatmap_1.0.13
# [79] memoise_2.0.1 Rcpp_1.1.1-1.1 fastmatch_1.1-8
# [82] svglite_2.2.2 xfun_0.57 pkgconfig_2.0.3